Communiqué de presse du 28 mars 2006 - Résultats de l’étude épidémiologique

Résultats des deux études menées sur la Pancréatite Chronique héréditaire depuis novembre 2004
études menées à l’initiative de l’Association des Pancréatites Chroniques Héréditaires (APCH) et financées par Solvay Pharma (partenaire et sponsor exclusif)
Auteurs : V. Rebours ; MC. Boutron-Ruault ; C. Férec; M. Schnee; F. Maire;
P. Hammel ; P. Ruszniewski; P Lévy
Résumé des études
La première étude portait sur les caractéristiques épidémiologiques de la PCH :
Une maladie génétique qui se déclare généralement dès l’enfance et dont le diagnostic n’est porté en moyenne qu’après 9 années malgré des symptômes extrêmement douloureux. Elle concerne au minimum 78 familles et 180 patients en sont atteints. Une maladie dont les manifestations ne semblent pas différentes de la pancréatite chronique d’origine alcoolique.
La deuxième étude portait sur le taux d’incidence de l’adénocarcinome pancréatique au cours de la PCH :
Les patients atteints ont 87 fois plus de risque de développer un adénocarcinome pancréatique (AP) que la population générale. Ce risque étant fortement corrélé au tabagisme, au diabète et à l’absence de pancréatite aigüe. L’AP est la première cause de décès de ces patients.
La PCH est une maladie génétique qui se caractérise par la destruction progressive du pancréas avec de lourdes conséquences sur la qualité de vie au quotidien des patients. Une meilleure connaissance de cette pathologie par le corps médical permettrait un diagnostic plus rapide et une meilleure prise en charge des symptômes et de la douleur pour tous les patients et particulièrement chez le jeune enfant.
L’APCH (Association des Pancréatites Chroniques Héréditaires), association loi 1901 à but non lucratif, s’est donné pour missions de faire connaître la PCH afin d’en faciliter le diagnostic surtout chez le jeune enfant ; de contribuer à l’effort de recherche et à l’amélioration des pratiques de soins et d’apporter une aide technique et morale aux familles.
L’APCH se veut active et prévoit dans les prochains mois :
- l’organisation de groupes de parole entre patients et médecins du conseil scientifique,
- des réunions informatives avec un médecin spécialisé dans la douleur,
- des réunions de patients.
Une brochure est également disponible à l’attention du corps médical et des familles sur simple demande
Son Conseil Scientifique:
Président d’Honneur, Pr. Louis Le Bodic, (+) (Nantes)
Président, Pr. Philippe Ruszniewski , Chef de Service de Gastro-Entérologie - Hôpital Beaujon (Clichy)
Pr. Marc Barthet, Service d’ Hépato-Gastro-Entérologie - Hôpital Nord (Marseille)
Pr. Louis Buscail, Service de Gastro-Entérologie et Nutrition - Hôpital Rangueil (Toulouse)
Pr. Claude Ferec, Chef de Service, Laboratoire de génétique moléculaire et épidémiologique - INSERM (Brest)
Pr. Olivier Goulet, Service de Soins Intensifs en Gastro-Entérologie , Hôpital Necker , Paris
Dr. Pascal HammeL, Président du Club Français du Pancréas (Paris)
Pr. Philippe Levy, Service de Gastro-Entérologie - Hôpital Beaujon (Clichy)
Pr. Christian Partensky, Service de Chirurgie Digestive - Hôpital Edouard Herriot (Lyon)
Dr. Matthieu Schnee , C.H.D (La Roche sur Yon )
Contact presse APCH : Nadine Meslet – Tel : 01 46 42 61 07 - E-mail : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Relations scientifiques : Pr Philippe Lévy – Hôpital Beaujon (Clichy) Tel : 01 40 87 52 15
Contact Solvay-Pharma : Dr François Dyard Tel 01 46 25 85 00 : E-mail : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Association loi 1901 J.O. 31 janvier 2004 . SIRET N°453 712 663 00013 . code APE 913 E
25 allée des Citeaux - 92130 Issy les Moulineaux
Tél. 01 46 42 61 07
e-mail : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
www.association-apch.org
Résultats des études présentées lors des JFPD (Journées de Pathologie digestive) Paris mars 2006
Résumé sélectionné (1ère étude)
Titre :
Histoire naturelle de la pancréatite chronique héréditaire (PCH) :
enquête nationale exhaustive
Auteurs/ Adresses :
V Rebours (1); MC Boutron-Ruault (2); C Férec (3); M Schnee (4); F Maire (1); P Hammel (1);
P Ruszniewski (1); P Lévy (1)
Association des Pancréatites Chroniques Héréditaires (APCH)
(1) Paris - FRANCE; (2) Villejuif - FRANCE; (3) Brest - FRANCE; (4) La-Roche-Sur-Yon - FRANCE;
Résumé :
La PCH est due à une mutation autosomique dominante du trypsinogène cationique (PRSS1). Sa prévalence et son histoire naturelle en France ne sont pas connues
Buts : Décrire les caractéristiques épidémiologiques, génétiques, cliniques et morphologiques de la PCH à partir d’une enquête exhaustive nationale.
Patients et Méthodes:
Une cohorte comprenant tous les pts atteints de PCH sur le territoire français a été constituée après avoir contacté tous les gastroentérologues et pédiatres français (taux de réponse : 84%) et les laboratoires de génétique (taux de réponse : 100%). Les critères d’inclusion étaient la présence d’une mutation du gène PRSS1 ou la présence d’une PC chez au moins deux apparentés au premier degré ou trois apparentés au second degré sans autre cause. Les mutations des gènes CFTR et de l’inhibiteur du trypsinogène de type 1 (SPINK1) étaient systématiquement cherchées.
Résultats
La cohorte comprenait 78 familles et 200 pts dont 181 vivants (nombre médian de génération par famille : 3, hommes : 53%, alcoolisme : 5%, tabagisme : 34%). Les mutations de PRSS1 étaient présentes ou absentes chez respectivement 68% et 32% (R122H : 78%, N29I : 12%, autres : 10%). De plus, les mutations de SPINK1 et CFTR étaient présentes chez respectivement 13% et 2% des pts. La transmission de PRSS1 était maternelle chez 65%. L’âge médian à l’inclusion et aux premiers symptômes était respectivement 30 ans (extrêmes : 1-84) et 10 ans (1-73). 17% des pts n’avaient aucun symptôme (pénétrance : 83%). Le délai médian entre les 1ers symptômes et le diagnostic était de 9 ans. Les manifestations étaient des douleurs pancréatiques (83% ; continues : 5%) ; une pancréatite aiguë (69%), un pseudokyste (23%), une cholestase (3%), des calcifications pancréatiques (61% ; âge médian d’apparition : 23 ans), une stéatorrhée (34%), un diabète (26%) et un adénocarcinome (5%, âge médian : 55 ans). L’âge médian de survenue de la stéatorrhée et du diabète était respectivement 29 et 38 ans. Il n’y avait pas de différence significative des données cliniques et morphologiques selon le statut génétique. Un traitement antalgique chronique (>3 mois) était nécessaire chez 10% des pts, un traitement endoscopique chez 16% et une intervention chirurgicale chez 23% (n = 49 dont 29 dérivations pancréatiques, 16 splénopancréatectomies caudales, 7 dérivations de pseudokyste, 4 duodéno-pancréatectomies céphaliques). Le nombre de décès était de 19 (dont 10 imputables à la PCH) survenant à l’âge médian de 60 ans.
Conclusion:
Le nombre de familles et de malades atteints de PCH en France peut être estimé au minimum à 78 familles et 180 malades. Une mutation de PRSS1 est trouvée dans 68% des cas avec une pénétrance de 83%. Il n’y a pas de différence clinique et morphologique selon le type de mutation. La PCH se déclare dès l’enfance, le diagnostic en est tardif. Les manifestations ne semblent pas différentes de celle de la PC alcoolique en dehors de la moindre fréquence de la cholestase et des insuffisances pancréatiques.
Résumé sélectionné (2 ème étude)
Titre :
Taux d'incidence standardisé de l'adénocarcinome pancréatique au cours de la pancréatite chronique héréditaire (PCH)
Auteurs :
V Rebours (1); MC Boutron-Ruault (2); C Férec (3); M Schnee (4); F Maire (1); P Hammel (1); P Ruszniewski (1); P Lévy (1); Association des Pancréatites Chroniques Héréditaires (APCH) (1) Paris – FRANCE (2) Villejuif – France (3) Brest – France (4) La-Roche-Sur-Yon – France
Mots clés : 27 Pancréatites Chroniques 70 Pancréas Exocrine
Résumé :
Introduction :
La PCH est due à une mutation autosomique dominante du trypsinogène cationique (PRSS1). La pancréatite chronique (PC) est un facteur de risque d’adénocarcinome pancréatique (AP) (SIR : 27) 1. La fréquence de l’AP au cours de la PCH est mal connue.
Buts :
Calculer l’incidence de l’AP et chercher ses facteurs de risque au cours de la PCH dans une série exhaustive nationale française.
Patients et Méthodes : Une cohorte comprenant tous les pts atteints de PCH sur le territoire français a été constituée après avoir contacté tous les laboratoires de génétique (taux de réponse : 100%) et les gastroentérologues et pédiatres français (taux de réponse : 84%). Les critères d’inclusion étaient la présence d’une mutation du gène PRSS1 ou la présence d’une PC chez au moins deux apparentés au premier degré ou trois apparentés au second degré sans autre cause. Les mutations des gènes CFTR et de l’inhibiteur du trypsinogène de type 1 (SPINK1) étaient systématiquement cherchées. Le diagnostic d’AP était toujours prouvé histologiquement et la cause de décès cherchée.
Résultats : La cohorte comprenait 78 familles et 200 pts dont 181 vivants représentant 6673 personnes-années (nombre médian de générations : 3, hommes : 53%, alcoolisme : 5%, tabagisme : 34%). L’âge médian à l’inclusion était 30 ans (extrêmes : 1-84). Les mutations de PRSS1 étaient présentes ou absentes chez respectivement 68% et 32% (R122H : 78%, N29I : 12%, autres : 10%). De plus, une mutation de SPINK1 et CFTR était présente chez respectivement 13% et 2% des pts. La transmission de PRSS1 était maternelle chez 65%. 10 AP ont été diagnostiqués (6 H, 4 F) à un âge médian de 55 ans (extrêmes : 39-78). Le SIR des AP pour l’ensemble de la cohorte, pour les hommes et pour les femmes était respectivement de 87 (IC 95% : 42-113), 69 (25-150) et de 142 (38-225). A 50, 60 et 75 ans, le risque cumulé d’avoir un AP était respectivement de 11%, 16% et 49% chez les hommes et 8%, 22% et 55% chez les femmes. Ce risque chez les fumeurs et les non fumeurs était respectivement de 72% et 16% à 75 ans. Le tableau compare les caractéristiques des malades avec ou sans AP:
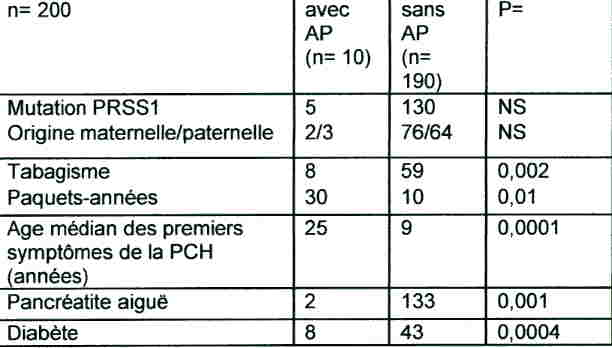
19 malades sont décédés dont 8 pts avec AP (42%, délai médian : 5 mois après le diagnostic). La cause du décès était connue chez tous les autres malades sauf 1.
Conclusion:
Les pts atteints de PCH ont 87 fois plus de risque de développer un AP que la population générale en France ; ceci peut être en partie imputable à la longue durée d’évolution de la PC. Ce risque n’est pas lié au statut génétique de PRSS1 mais est fortement corrélé au tabagisme, au diabète et à l’absence de pancréatite aiguë. L’AP la première cause de décès de ces patients.Résultats des deux études menées sur
la Pancréatite Chronique héréditaire depuis novembre 2004
études menées à l’initiative de l’Association des Pancréatites Chroniques Héréditaires (APCH) et
financées par Solvay Pharma (partenaire et sponsor exclusif)
Auteurs : V. Rebours ; MC. Boutron-Ruault ; C. Férec; M. Schnee; F. Maire;
P. Hammel ; P. Ruszniewski; P Lévy